Macaques and sample size
Indian-origin rhesus macaques (Macaca mulatta) used in these studies are outlined in Extended Data Fig. 1c and Supplementary Table 1. All experimentation complied with ethical regulations at the respective institutions (Animal Care and Use Committees of the Vaccine Research Center, NIAID, NIH and of Bioqual, Inc., and of the Institutional Animal Care and Use Committee of the University of Pittsburgh). Macaques were housed and cared for in accordance with local, state, federal, and institute policies in facilities accredited by the American Association for Accreditation of Laboratory Animal Care (AAALAC), under standards established in the Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals. Macaques were monitored for physical health, food consumption, body weight, temperature, complete blood counts, and serum chemistries. All infections were performed at the University of Pittsburgh where animals were housed in a biosafety level 3 facility.
The sample size for this study was determined using bacterial burden (measured as log10-transformed total thoracic CFUs) as the primary outcome variable. Initially, we planned to test BCG route efficacy by comparing IV, AE and AE/ID routes to IDlow vaccination and found that ten macaques per group would be sufficient to obtain over 90% power and adjusted the type I error rate for three group comparisons (α = 0.0167). After initiation of the first cohort of NHPs in this study, we elected to test the effect of dose on ID vaccination by adding an IDhigh group (n = 8 macaques). The additional treatment group did not substantially reduce the power of the study. To detect a 1.5 difference in log10(total CFUs) with a pooled standard deviation of 0.8 (using previous data), we obtained over 90% (90.7%) power using 10 macaques per group with an adjusted type I error rate for 4 group comparisons (α = 0.0125). The comparison made between the IDhigh (n = 8 macaques) and IDlow (n = 10 macaques) groups achieved 85.6% power detecting the same difference (log10(1.5)) and with an α = 0.0125.
BCG vaccination
For Mtb challenge studies (cohorts 1–3), 3–5-year-old male (n = 32) and female (n = 20) rhesus macaques were randomized into experimental groups based on gender, weight and pre-vaccination CD4 T cell responses to PPD in BAL. Macaques were vaccinated at Bioqual, Inc. under sedation and in successive cohorts as outlined in Extended Data Fig. 1c. BCG Danish Strain 1331 (Statens Serum Institute, Copenhagen, Denmark) was expanded42, frozen at approximately 3 × 108 CFUs ml−1 in single-use aliquots and stored at −80 °C. Immediately before injection, BCG (for all vaccine routes) was thawed and diluted in cold PBS containing 0.05% tyloxapol (Sigma-Aldrich) and 0.002% antifoam Y-30 (Sigma-Aldrich) to prevent clumping of BCG and foaming during aerosolization43. For ID vaccinations, BCG was injected in the left upper arm (5 × 105 CFUs; IDlow) or split across both upper arms (5 × 107 CFUs; IDhigh) in a volume of 100–200 μl per site. IV BCG (5 × 107 CFUs) was injected into the left saphenous vein in a volume of 2 ml; AE BCG (5 × 107 CFUs) was delivered in a 2 ml volume via paediatric mask attached to a Pari eFlow nebulizer (PARI Pharma GmgH) that delivered 4 μM particles into the lung, as previously described28; AE/ID macaques were immunized simultaneously (5 × 107 CFUs AE plus 5 × 105 CFUs ID in left arm); EB BCG (5 × 107 CFUs in 2 ml; cohort 6 only) was instilled into the left lung lobes using an endoscope. No loss of viability was observed for BCG after aerosolization. In pilot studies, lower doses of BCG were prepared and delivered as described above. Text refers to nominal BCG doses—actual BCG CFUs for vaccine regimens in every cohort were quantified immediately after vaccination and are reported in Extended Data Fig. 1c and Supplementary Table 1.
Mtb challenge
Macaques (cohorts 1–3) were challenged by bronchoscope with 4–36 CFUs barcoded Mtb Erdman 6–10 months after BCG vaccination (Extended Data Fig. 1c and Supplementary Table 1) in a 2 ml volume as previously described44. Infectious doses across this range result in similar levels of TB disease in unvaccinated rhesus in this and previous studies28 (Supplementary Data 12). Clinical monitoring included regular monitoring of appetite, behaviour and activity, weight, erythrocyte sedimentation rate, Mtb growth from gastric aspirate and coughing. These signs, as well as PET–CT characteristics, were used as criteria in determining whether a macaque met the humane end point before the pre-determined study end point.
PET–CT scans and analysis
PET–CT scans were performed using a microPET Focus 220 preclinical PET scanner (Siemens Molecular Solutions) and a clinical eight-slice helical CT scanner (NeuroLogica Corporation) as previously described27,45,46,47. 2-deoxy-2-(18F)fluorodeoxyglucose (FDG) was used as the PET probe. Serial scans were performed before, 4 and 8 weeks after Mtb, and before necropsy (cohorts 1–3) or at 2 and 4 weeks after BCG (cohorts 5a, b). OsiriX MD (v.10.0.1), a DICOM (Digital Imaging and Communications in Medicine) image viewer, was used for scan analyses, as described47. Lung inflammation was measured as total FDG activity within the lungs. A region of interest (ROI) was segmented which encompassed all lung tissue on CT and was then transferred to the co-registered PET scan. On the PET scan, all image voxels of FDG-avid pathology (Standard Uptake Value >2.3) were isolated and summated resulting in a cumulative standardized uptake value. To account for basal metabolic FDG uptake, total FDG activity was normalized to resting muscle resulting in a total lung inflammation value. Individual granulomas were counted on each CT scan. If granulomas were too small and numerous within a specific area to count individually or if they consolidated, quantification was considered to be too numerous to count. To measure the volume of the spleen, an ROI was drawn outlining the entire organ on each of the axial slices of the CT scan and the volume was computed across these ROIs (using a tool in OsiriX). Any scans for which visibility of the entire spleen was limited (n = 2 macaques) were excluded from this analysis.
Necropsy, pathology scoring and Mtb and BCG burden
For challenge studies (cohorts 1–3), NHPs were euthanized 11–15 weeks after Mtb or at humane endpoint by sodium pentobarbital injection, followed by gross examination for pathology. A published scoring system27 was used to determine total pathology from each lung lobe (number and size of lesions), LN (size and extent of necrosis), and extrapulmonary compartments (number and size of lesions). All granulomas and other lung pathologies, all thoracic LNs, and peripheral LNs were matched to the final PET–CT scan and collected for quantification of Mtb. Each lesion (including granulomas, consolidations and clusters of granulomas) in the lung, all thoracic LNs, random sampling (50%) of each of the 7 lung lobes, 3–5 granulomas (if present) or random samples (30%) of spleen and liver, and any additional pathologies were processed to comprehensively quantify bacterial burdens. Suspensions were plated on 7H11 agar (Difco) and incubated at 37 °C with 5% CO2 for 3 weeks for CFU enumeration or formalin-fixed and paraffin-embedded for histological examination. CFUs were counted and summed to calculate the total thoracic bacterial burden for the macaque17,27,48. Mtb CFUs for every challenged macaque are listed in Supplementary Table 1.
To determine BCG CFUs, BAL, bone marrow aspirates, and blood were collected from NHPs before euthanasia. Individual lung lobes and thoracic and peripheral LNs, spleen, liver, and the skin site(s) of injection (if applicable) were excised. 0.5 ml of blood and bone marrow and 10% of retrieved BAL wash fluid were plated; approximately 1 g of tissue (or one whole LN or skin biopsy) was processed in water in gentleMACS M Tubes (Miltenyi Biotec) using a gentleMACS Dissociator (Miltenyi Biotec). Samples were plated and counted as above. Data are reported as CFUs ml−1 of blood or bone marrow, CFUs per total BAL collected, CFUs per one LN or skin biopsy, CFUs per lung lobe or spleen. CFUs from individual lung lobes and LNs of the same category (for example, hilar) were averaged for each NHP.
Rhesus blood, BAL and tissue processing
Blood PBMCs were isolated using Ficoll-Paque PLUS gradient separation (GE Healthcare Biosciences) and standard procedures; BAL wash fluid (3 × 20 ml washes of PBS) was centrifuged and cells were combined before counting, as described28. LNs were mechanically disrupted and filtered through a 70-μm cell strainer. Lung and spleen tissues were processed using gentleMACS C Tubes and Dissociator in RPMI 1640 (ThermoFisher Scientific). Spleen mononuclear cells were further separated using Ficoll-Paque. Lung tissue was digested using collagenase, Type I (ThermoFisher Scientific) and DNase (Sigma-Aldrich) for 30–45 min at 37 °C with shaking, followed by passing through a cell strainer. Single-cell suspensions were resuspended in warm R10 (RPMI 1640 with 2 mM l-glutamine, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, and 10% heat-inactivated FBS; Atlantic Biologicals) or cryopreserved in FBS containing 10% DMSO in liquid nitrogen.
Multiparameter flow cytometry
Generally, longitudinal PBMC samples were batch-analysed for antigen-specific T cell responses or cellular composition at the end of the study from cryopreserved samples whereas BAL and tissue (necropsy) samples were analysed fresh. Cryopreserved PBMC were washed, thawed and rested overnight in R10 before stimulation, as described28. For T cell stimulation assays, 1–5 million viable cells were plated in 96-well V-bottom plates (Corning) in R10 and incubated with R10 alone (background), or with 20 μg ml−1 tuberculin PPD (Statens Serum Institut, Copenhagen, Denmark), 20 μg ml−1 H37Rv Mtb WCL (BEI Resources), or 1 μg ml−1 each of ESAT-6 and CFP-10 peptide pools (provided by Aeras, Rockville, MD) for 2 h before adding 10 μg ml−1 BD GolgiPlug (BD Biosciences). The concentrations of PPD and WCL were optimized to detect CD4 T cell responses; however, protein antigen stimulation may underestimate CD8 T cell responses. For logistical reasons, cells were stimulated overnight (14 h total) before intracellular cytokine staining. For cellular composition determination, cells were stained immediately ex vivo after processing or after thawing. Antibody and tetramer information for each flow cytometry panel is listed in Supplementary Data 8–11. Generally, cells were stained as follows (not all steps apply to all panels, all are at room temperature): Washed twice with PBS/BSA (0.1%); 20-min incubation with rhesus MR1 tetramer49 (NIH Tetramer Core Facility) in PBS/BSA; washed twice with PBS; live/dead stain in PBS for 20 min; washed twice with PBS/BSA; 10-min incubation with human FcR blocking reagent (Miltenyi Biotec); incubation with surface marker antibody cocktail in PBS/BSA containing 1× Brilliant Stain Buffer Plus (BD Biosciences) for 20 min; washed three times with PBS/BSA (0.1%); 20 min incubation BD Cytofix/Cytoperm Solution (BD Biosciences); washed twice with Perm/Wash Buffer (BD Biosciences); 30 min incubation with intracellular antibody cocktail in Perm/Wash Buffer containing 1× Brilliant Stain Buffer Plus; washed thrice with Perm/Wash Buffer. For Ki-67 staining, samples were stained for surface markers and cytokines as described above, followed by nuclear permeabilization using eBioscience Foxp3/Transcription Factor Staining Buffer (ThermoFisher Scientific) and incubation with antibody against Ki-67 following kit instructions. Data were acquired on either a modified BD LSR II or modified BD FACSymphony and analysed using FlowJo software (v.9.9.6 BD Biosciences). Gating strategies can be found in Supplementary Data 8–11. All cytokine data presented graphically are background-subtracted.
Intravascular CD45 staining
One month after BCG vaccination, macaques in each cohort 6 (n = 2 macaques per group) received an IV injection of Alexa Fluor 647-conjugated anti-CD45 antibody (ivCD45; 60 μg kg−1, clone MB4-6D6, Miltenyi Biotec) 5 min before euthanasia. Blood was collected before anti-CD45 injection as a negative control, and before euthanasia as a positive control. NHPs underwent whole body perfusion with cold saline before tissue collection. Tissues were processed for BCG CFU quantification and flow cytometric analysis as described above. Staining panels used were as in Supplementary Data 9, with the omission of the APC-conjugated antibodies.
Immunohistochemistry
Embedded tissue sections were deparaffinized (100% xylenes, 10 min; 100% ethanol, 5 min; 70% ethanol, 5 min), boiled under pressure for 6 min in antigen retrieval buffer (1× Tris EDTA, pH 9.0), and cooled. Sections were blocked in PBS (1% BSA) in a humidified chamber at room temperature for 30 min followed by staining for CD3 (CD3-12, Abcam), CD11c (5D11, Leica), and CD20 (Thermo Scientific, RB-9013-PO) for 18 h at 4 °C in a humidified chamber. After washing with PBS in coplin jars, sections were incubated for 1 h at room temperature with conjugated anti-rabbit IgG Alexa Fluor 488 (Life Technologies, A21206), anti-rat IgG Alexa Fluor 546 (Invitrogen, A11081), and anti-mouse IgG Alexa Fluor 647 (Jackson ImmunoResearch, 7 5606-150). After washing, coverslips were applied using Prolong Gold anti-fade with Dapi mounting media (Life Technologies). Slides were cured for 18–24 h before imaging on an Olympus FluoView FV1000 confocal microscope. Lung sections were imaged and two random representative 1 mm2 ROIs from each macaque were analysed using CellProfilerv2.2.0. Pipelines were designed for analysis by adding modules for individual channel quantification based on pixel intensity and pixel size providing a numerical value for each cell type and total cells. Histological analyses were performed by a veterinary pathologist (E.K.) in a blinded fashion on H&E-stained sections from all tissues obtained.
ELISpot and Luminex
IFNγ ELISpots were performed at 0, 4, 6 and 8 weeks after Mtb and at necropsy. One day before use, hydrophobic high protein binding membranes 96-well plates (Millipore Sigma) were hydrated with 40% ethanol, washed with sterile water, and coated with anti-human/monkey IFNγ antibody (15 μg ml−1, MT126L, MabTech) overnight at 4 °C. Plates were washed with HBSS and blocked with RPMI with 10% human AB serum for 2 h at 37 °C with 5% CO2. Approximately 200,000 PBMCs per well were incubated in RPMI supplemented with l-glutamate, HEPES and 10% human AB serum containing 2 μg ml−1 ESAT-6 or CFP-10 peptide pools for 40–48 h at 37 °C with 5% CO2. Medium alone or phorbol 12,13-dubutyrate (12.5 μg ml−1) plus ionomycin (37.5 μg ml−1) were added as negative (background) and positive controls, respectively. To develop, plates were washed with PBS and biotinylated anti-human IFNγ antibody (2.5 μg ml−1, 7-B6-1, MabTech) was added for 2 h at 37 °C with 5% CO2. After washing, streptavidin-horseradish peroxidase (1:100, MabTech) was added for 45 min at 37 °C with 5% CO2. Spots were stained using AEC peroxidase (Vector Laboratories, Inc.) per the manufacturer’s instructions and counted manually on an ELISpot plate reader. Data are reported as average ELISpots from duplicate background-subtracted wells. Wells with confluent spots were described as too numerous to count.
To measure innate cytokine production following BCG immunization, cryopreserved PBMC were batch-analysed. Cells were thawed and resuspended in warm R10. Then, 5 × 105 cells per well in 96-well V-bottom plates were rested overnight at 37 °C with 5% CO2. Cells were resuspended in Trained Immunity Media15 plus H37Rv Mtb whole cell lysate (BEI Resources, 20 μg ml−1), heat-killed Staphylococcus aureus (InvivoGen, 1 × 106 per ml), Escherichia coli LPS (Sigma-Aldrich, 1 ng ml−1), or RPMI and incubated for 24 h at 37 °C with 5% CO2 before collecting supernatants. Cytokine and chemokine measurements were determined using a MILLIPLEX NHP cytokine multiplex kit per instructions (Millipore Sigma) and analysed on a Bio-Plex Magpix Multiplex Reader (Bio-Rad).
Antibody ELISAs
IgG, IgA and IgM titres to Mtb H37Rv WCL were assessed in plasma and tenfold concentrated BAL fluid. WCL was used based on greater sensitivity compared to PPD, culture filtrate protein, or lipoarabinomannan. 96-well MaxiSorp ELISA plates (Nunc) were coated overnight at 4 °C with 0.1 μg of WCL. Plates were blocked with PBS/FBS (10%) for 2 h at room temperature and washed with PBS/TWEEN 20 (0.05%). 1:5 serially diluted plasma or concentrated BAL fluid (8 dilutions per sample) was incubated at 37 °C for 2 h, followed by washing. Then, 100 μl of goat anti-monkey HRP-conjugated IgG h+l (50 ng ml−1; Bethyl Laboratories, Inc.), IgA α chain (0.1 μg ml−1, Rockland Immunochemicals Inc.), or IgM α chain (0.4 μg ml−1, Sera Care) was added for 2 h at room temperature, followed by washing. Ultra TMB substrate (100 μl; Invitrogen) was added for 12 min followed by 100 μl 2 N sulfuric acid. Data were collected on a Spectramax i3X microplate reader (Molecular Devices) at 450 nm using Softmax Pro and presented either as endpoint titer (reciprocal of last dilution with an OD above the limit of detection or 2× the OD of an empty well) at 0.2 for IgG and IgA, or midpoint titer for IgM where samples did not titre to a cut off of 0.2.
Single-cell transcriptional profiling
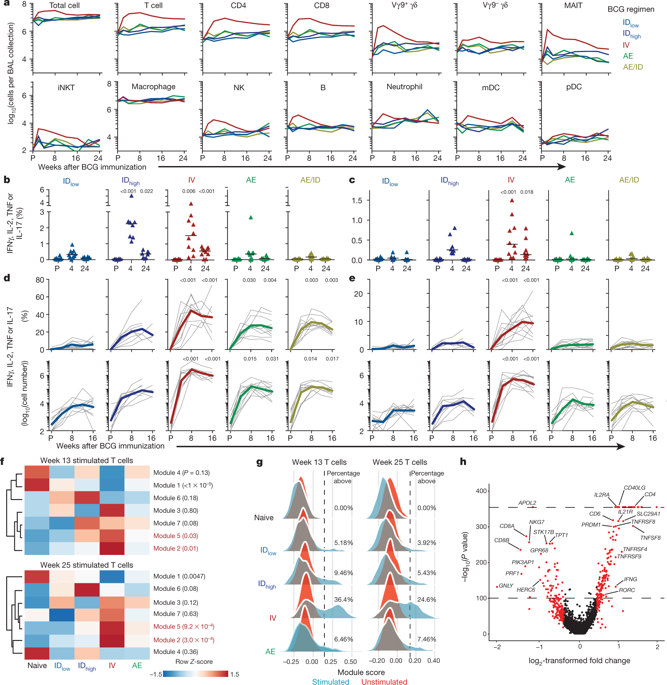
High-throughput single-cell mRNA sequencing by Seq-Well was performed on single-cell suspensions obtained from NHP BAL, as previously described24. Approximately 15,000 viable cells per sample were applied directly to the surface of a Seq-Well device. At each time point after BCG, two arrays were run for each sample—one unstimulated and one stimulated overnight with 20 μg ml−1 of PPD in R10.
Sequencing and alignment
Sequencing for all samples was performed on an Illumina Nova-Seq. Reads were aligned to the M. mulatta genome using STAR50, and the aligned reads were then collapsed by cell barcode and unique molecular identifier (UMI) sequences using DropSeq Tools v.1 to generate digital gene expression (DGE) matrices, as previously described24,51. To account for potential index swapping, we merged all cell barcodes from the same sequencing run that were within a hamming distance of 1.
Analysis of single-cell sequencing data
For each array, we assessed the quality of constructed libraries by examining the distribution of reads, genes and transcripts per cell. For each time point, we next performed dimensionality reduction (PCA) and clustering as previously described52,53. We visualized our results in a two-dimensional space using UMAP54, and annotated each cluster based on the identity of highly expressed genes. To further characterize substructure within cell types (for example, T cells), we performed dimensionality reduction (PCA) and clustering over those cells alone as previously described24. We then visualized our results in two-dimensional space using t-distributed stochastic neighbour embedding (t-SNE)24. Clusters were further annotated (that is, as CD4 and CD8 T cells) by cross-referencing cluster-defining genes with curated gene lists and online databases (that is, SaVanT andGSEA/MsigDB)55,56,57.
Module identification
Data from stimulated or unstimulated T cells at week 13 or 25 was subset on significant principal components as previously described24 and, for those principal components, on genes with significant loadings as determined through a randomization approach (‘JackStraw’)52. These matrices were then used as the inputs for WGCNA58. Following the WGCNA tutorial (https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/Tutorials/), we chose an appropriate soft power threshold to calculate the adjacency matrix. As scRNA-seq data is affected by transcript drop-out (failed capture events), adjacency matrices with high power further inflate the effect of this technical limitation, and yield few correlated modules. Therefore, when possible, we chose a power as suggested by the authors of WGCNA (that is, the first power with a scale free topology above 0.8); however, if this power yielded few modules (fewer than three), we decreased our power. We then generated an adjacency matrix using the selected soft power and transformed it into a topological overlap matrix (TOM). Subsequently, we hierarchically clustered this TOM, and used the cutreeDynamic function with method ‘tree’ to identify modules of correlated genes using a dissimilarity threshold of 0.5 (that is, a correlation of 0.5). To test the significance of the correlations observed in each module, we implemented a permutation test. Binning the genes in the true module by average gene expression (number of bins = 10), we randomly picked genes with the same distribution of average expression from the total list of genes used for module discovery 10,000 times. For each of these random modules, we performed a one-sided Mann–Whitney U-test between the distribution of dissimilarity values among the genes in the true module and the distribution among the genes in the random module. Correcting the resulting P values for multiple hypothesis testing by Benjamini–Hochberg false discovery rate correction, we considered the module significant if fewer than 500 tests (P < 0.05) had false discovery rate > 0.05.
Gene module enrichments
To characterize the seven significant gene modules identified among in vitro-stimulated T cells collected 13 weeks after vaccination, we performed an enrichment analysis using databases of gene expression signatures (SaVanT and GSEA/MsigDb). Specifically, the enrichments in the Savant database, which includes signatures from ImmGen, mouse body atlas and other datasets (http://newpathways.mcdb.ucla.edu/savant-dev/), were performed using genes included in significant modules with a background expression set of 32,681 genes detected across single cells using Piano (https://varemo.github.io/piano/).
Statistical methods
All reported P values are from two-sided comparisons. For continuous variables, vaccine routes were compared using a Kruskal–Wallis test with Dunn’s multiple comparison adjustment or one-way ANOVA with Dunnett’s multiple comparison adjustment (comparing all routes to IDlow BCG). Fisher’s exact tests were run for multiple CFU thresholds (evaluating protection) to assess the association between vaccine route and protection from Mtb (Extended Data Fig. 8b). A permutation test59 was used to compare fractional distributions (pie charts) of all vaccine groups to IDlow BCG. For clinical parameters, combined pre-vaccination measurements from all NHPs were compared against distributions from every vaccine group at every time point using Dunnett’s test for multiple comparisons. To assess whether post-vaccination antigen-responsive CD4 or CD8 T cells in the BAL or PBMCs are associated with disease severity, we first calculated peak T cell responses for each NHP over the course of vaccine regimen. The log10-transformed CD4 and CD8 cell counts were calculated within BAL and frequencies of CD4 and CD8 cells were calculated within PBMCs. To assess the effects of vaccine route and T cells on log10-transformed total CFUs, several multiple linear regressions were run in JMP Pro (v.12.1.0). Peak T cell responses and CFUs for each macaque included in these analyses are provided in Supplementary Table 1; detailed regression output (including model fit, ANOVA results, effect tests and parameter estimates) is provided in Supplementary Table 5. Cytokine production for trained immunity assay was compared using a two-way ANOVA and Dunnett’s multiple comparison test. Serial PBMC responses to CFP, ESAT-6 or CFP-10 by IFNγ ELISpot were analysed by using a Wilcoxon signed-rank test to compare pre-infection versus 12 weeks post-infection time points (within each vaccine route).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.